Qué es un ensayo clínico

Un ensayo clínico es una investigación médica que se realiza para descubrir los efectos clínicos y farmacológicos que los medicamentos producen en las personas. Así como para identificar sus reacciones adversas. Y, pese a que se practican también con otros fines, los más habituales son los que se utilizan para evaluar nuevos fármacos y procedimientos que todavía no están establecidos. Precisamente dos áreas en las que los pacientes nos encontramos en un momento de mucha vulnerabilidad y miedo a la incertidumbre.
Aun así, es necesario destacar que es una práctica regulada que se lleva a cabo bajo las máximas garantías éticas, regulatorias y médicas. De hecho, no todas las propuestas que se exploran terminan probándose en seres humanos. Los nuevos fármacos deben analizarse primero en un laboratorio para que, una vez identificados los prometedores, pasen a ser estudiados con más profundidad en los ensayos clínicos; la etapa del I+D farmacéutico en la que los pacientes nos convertimos, ya como usuarios, en la máxima prioridad. La participación en los ensayos debe ser además voluntaria, aunque si que está condicionada al cumplimiento de unos requisitos preestablecidos y a la aceptación expresa de los términos recogidos en el “consentimiento informado”. Pese a esto, se puede cambiar de opinión y salirnos en cualquier momento de la investigación, sin que esta decisión nos condicione a la asistencia médica posterior.
La voluntad de los pacientes se respeta y los datos se tratan en todo momento con rigurosa privacidad y confidencialidad.
La primera información que se explica sobre los ensayos clínicos es que tienen como objetivo principal comprobar la seguridad y eficacia de los tratamientos a estudiar. Es decir, que los pacientes no nos veamos perjudicados al participar. Y sin renunciar nunca a este aspecto, comprobar al mismo tiempo los efectos en la salud que estos puedan ocasionarnos
Siempre con la finalidad de ayudarnos a los participantes y a futuros pacientes en la misma situación. De ahí que se comience en un entorno controlado bien definido que se amplía según se genera la evidencia que garantiza que el tratamiento funciona y es seguro. Todos los fármacos disponibles en la actualidad han sido cuidadosamente testados con esta metodología. La única, después de todo, que permite las partes implicadas poder comparar y tomar decisiones: la administración pública para decidir si autoriza o no ese fármaco. Los médicos para determinar qué tratamientos son los más adecuados para sus pacientes. Y, en nuestro caso, para que podamos tomar la decisión final de aceptar la propuesta que más se ajuste a los beneficios y riesgos que estemos dispuestos a asumir.
Sin embargo, no todos los ensayos clínicos logran su propósito. Un fármaco experimental podría resultar ser menos eficaz que otros tratamientos habituales que ya se están empleando en la práctica clínica. O puede ser que sus efectos secundarios sean más frecuentes o severos que los que presentan otras alternativas. Pero, incluso así, esta información seguiría siendo valiosa y podría resultar clave para futuras investigaciones.
Al fin y al cabo, son los investigadores (médicos y/u otros profesionales sanitarios) expertos en el diagnóstico y tratamiento de pacientes con una enfermedad concreta los que diseñan los ensayos clínicos. Y el conocimiento se acumula, tanto para idear nuevos acercamientos con los que abordar la misma enfermedad, como para que las autoridades competentes evalúen y admitan estas propuestas. En España estas funciones recaen en los Comités de Ética y en la Agencia Española de Medicamentos y Productos Sanitarios (AEMPS), adscrita al Ministerio de Sanidad, Consumo y Bienestar Social. Una labor que realizan analizando los detalles que se especifican en el protocolo. Un documento que, cumpliendo con la normativa ética y legal, hay que seguir de forma estricta porque es lo que contiene lo que se va a hacer, por qué, cómo, con quién y los tiempos de actuación. Y esto es lo que se aprueba, por lo que no se puede alterar. Es aquí donde también se explican los requisitos que hemos de cumplir los participantes. Si bien, el criterio que prevalece, junto al de voluntariedad de la participación, es el de la seguridad de los pacientes.
Proceso de un ensayo clínico
Proceso de un ensayo clínico
Diseño de un ensayo clínico
Diseño de un ensayo clínico

Los ensayos con humanos se plantean cuando finaliza la investigación básica y preclínica, las fases en las que se realizan la experimentación en cultivos de células (in vitro) y con animales.
Es precisamente en este punto cuando se lleva a cabo el diseño del ensayo clínico, con tres parámetros fundamentales a definir:
- 1. Los objetivos de estudio, que son de dos tipos: primarios y secundarios. El objetivo primario es con el que los investigadores determinan que el nuevo fármaco se puede considerar beneficioso para las personas. Los objetivos secundarios reportan un beneficio adicional. En la mayoría de los estudios se considera la eficacia como el objetivo principal.
- 2. La población a estudiar, lo que se hará en función de los objetivos fijados. Debe de guardar coherencia con la población a la que luego, una vez aprobado el fármaco, se quiere destinar. Por ello, se establecerán criterios de inclusión y exclusión de participantes. Además, se deberá hacer una selección de una muestra representativa, ya que es necesario saber el número mínimo de personas que se deben incluir para que el ensayo sea válido. Un aspecto que dependerá también de las variables a estudiar.
- 3. La dosis a emplear, que no debe ser ni excesivamente alta para evitar poner en riesgo la seguridad de los pacientes, ni excesivamente baja como para que no se pueda evaluar su eficacia.
Todos estos parámetros se reflejarán en el protocolo del ensayo clínico
Características de ensayos clínicos
Características de ensayos clínicos
De acuerdo con los estándares actuales de investigación científica se considera que los ensayos más completos y exhaustivos deberían de cumplir una serie de características. Algunas de ellas son:
- Utilizar un grupo de control:
Los ensayos controlados son aquellos en los que se realiza una comparación entre dos grupos: uno, que recibe la molécula en experimentación, y el otro que recibe placebo o un fármaco ya conocido. Este último grupo es el que se conoce como grupo de control. - Asignar aleatoriamente los participantes a los grupos:
Esto significa que los participantes forman parte de un grupo u otro del ensayo mediante una asignación aleatoria, teniendo igual probabilidad estadística de pertenecer a un grupo o al otro. - Aplicar el doble ciego:
Los ensayos que utilizan el doble ciego son aquellos en los que ni el participante ni el investigador conocen qué tratamiento es el que se está utilizando en cada caso particular. La finalidad es evitar el efecto placebo, así como cualquier sesgo en el investigador.
Fase previa
Fase previa
Creación del protocolo
Las características de un ensayo clínico se recogen en el protocolo, que es el documento que incluye la justificación y los objetivos del experimento; el diseño y la metodología a seguir; el procedimiento para el análisis previo de los resultados, así como las condiciones bajo las que se desarrollará el ensayo.
En el protocolo se especifica también tanto el tipo como el número de participantes que, como mínimo, han de incluirse para poder conseguirse el objetivo propuesto. Incluso se desglosan en él los criterios de inclusión y exclusión que han de seguirse para la admisión. Del mismo modo que se incluye toda la información relativa a los fármacos que serán utilizados, como la dosis, vía de administración, pauta y periodo de tratamiento.
El ensayo deberá realizarse de acuerdo con lo recogido en el protocolo una vez que es autorizado por las autoridades sanitarias. La realización de este documento la lleva a cabo un equipo multidisciplinar, por iniciativa del promotor del ensayo clínico.
Selección de los centros hospitalarios
El promotor es el agente encargado de seleccionar el/los centros hospitalarios donde se llevarán a cabo los ensayos clínicos.
Evaluación por parte del Comité Ético de Investigación Clínica
La aprobación del Comité Ético de Investigación Clínica es necesaria para que se lleve a cabo el ensayo. Ellos evalúan los aspectos legales, metodológicos y éticos que se seguirán durante esta fase de desarrollo del fármaco.
Autorización de la Agencia Española del Medicamento y Productos Sanitarios
Firmados los documentos de aprobación del protocolo por el investigador principal, el/los directores de los centros hospitalarios y el Comité Ético de Investigación Clínica, el promotor hace llegar la solicitud formal a la Agencia Española del Medicamento y Productos Sanitarios (AEMPS) para obtener la aprobación. Sin esa aprobación no puede comenzar el ensayo.
Fase de ejecución y desarrollo
Fase de ejecución y desarrollo
Tras la notificación y aprobación por las autoridades sanitarias, se pone en marcha la ejecución del ensayo clínico. Los centros hospitalarios participantes recibirán el material necesario para llevarlo a cabo, para que luego ellos puedan proceder con el reclutamiento de los participantes, una tarea que es dirigida por el investigador principal.
Es entonces cuando comienza lo que es el ensayo clínico en sí, que es la parte más extensa y compleja. Por ello, el monitor realizará una supervisión constante, para que se sigan correctamente las directrices fijadas en el protocolo. Además, irá recogiendo progresivamente los datos correspondientes a cada participante, hasta que se finalice el ensayo.
Los resultados
Los resultados
Tras recabar los datos de todos los participantes en el ensayo clínico, comienza la fase de análisis de dichos datos, en los que se contará con la ayuda de un equipo de profesionales expertos en estadística. Los resultados del análisis estadístico se publicarán en revistas científicas para darlos a conocer, de hecho, un ensayo no debe considerarse finalizado si no es publicado. Posteriormente, los resultados se recogerán en un informe que se adjuntará a la documentación necesaria para la aprobación del fármaco en experimentación.
Fases de la Investigación
Fases de la Investigación
Las fases de investigación de un fármaco antes de su aprobación son tres: investigación básica o fase de descubrimiento, ensayos preclínicos y ensayos clínicos, aunque solo en la última fase, en la de los ensayos clínicos, es en la que se realiza la investigación en humanos.
Fase de descubrimiento o de investigación básica
Fase de descubrimiento o de investigación básica
Es el primer paso que inicia todo el proceso, es decidir, en función de intereses científicos o estratégicos, la enfermedad sobre la que se quiere investigar con el fin de encontrar un medicamento que ayude a su prevención, tratamiento o mejora sobre los efectos que produce en el cuerpo. Una vez fijado el objetivo, se pasa a buscar moléculas que puedan influir beneficiosamente en la enfermedad escogida. Y esta búsqueda puede darse de dos maneras:
- Conociendo cómo funciona la enfermedad y los mecanismos biológicos implicados, se buscan moléculas que alteren la función del organismo, lo que se conoce como abordaje biológico.
- Partiendo de una serie de compuestos químicos previamente disponibles hasta observar cuáles de ellos tienen un efecto sobre el desarrollo de la misma: abordaje químico.
Esta fase es un proceso largo y lento. De forma aproximada, se estima que, por cada 10.000 moléculas en la etapa de investigación básica, sólo 250 entrarán en la siguiente etapa de investigación preclínica..
Fase pre-clínica
Fase pre-clínica
Las moléculas “líderes” de la primera fase pasaran ahora un nuevo test compuesto de dos partes. La primera y la más importante de todas es demostrar en el laboratorio –bien en animales, bien en cultivos de células- el perfil de seguridad de la molécula, comprobando cuáles son los potenciales efectos tóxicos y adversos que podrían causar en los seres humanos. Por ello, se estudia cómo funciona a distintas dosis, en distintos órganos y sistemas y cómo es eliminado por el cuerpo.
El fin último es que, desde esta fase del desarrollo de un fármaco, se preserve la seguridad de los pacientes, evitando posibles riesgos futuros.
La segunda parte de la prueba es comprobar la eficacia que podría tener a futuro antes de comenzar los ensayos en humanos, evaluando el mecanismo de acción de la molécula.
Con los resultados que se obtienen de esta fase se elabora un informe que se presenta a las autoridades sanitarias pertinentes para que den el visto bueno a continuar con la investigación y pasar a realizar ensayos clínicos, donde ya se realizan pruebas en personas.
Esta fase puede llegar a durar tres años o más y hay miles de compuestos que nunca pasan a la siguiente fase. Se estima que, por cada 250 compuestos en preclínica, sólo cinco entrarán en la siguiente etapa de investigación clínica.
Fase clínica
Fase clínica
La entidad que desarrolla el fármaco deberá presentar el informe con los resultados de la fase preclínica, además del plan detallado de cómo van a ser los estudios clínicos: número de participantes, descripción detallada, centros implicados, criterio de elección de pacientes, medidas de seguridad, etc. Es lo que se conoce como protocolo. Además, los ensayos serán supervisados por Comités Éticos de Investigación Clínica para garantizar los derechos de los pacientes.
Clasificación
Se clasifican en fases dependiendo de lo avanzada que esté la investigación. Un tratamiento no puede pasar a la siguiente fase si no se obtienen resultados satisfactorios en la fase en la que se encuentra. Estas fases son:
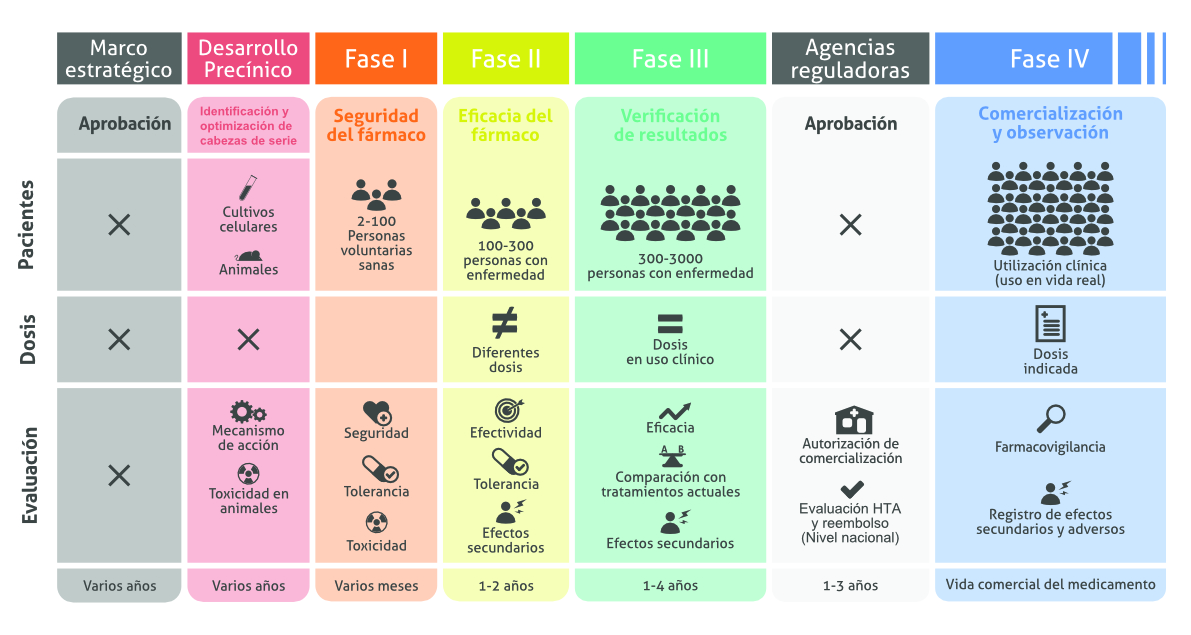
- Fase I: El fármaco se administra por primera vez en humanos. Las personas participantes debemos ser pacientes con diferentes tipos de cáncer para observar el efecto del fármaco, especialmente la seguridad. Dura entre seis meses y un año y en ella se realizan los primeros estudios en personas – entre 20 y 100 voluntarios pacientes – para ratificar la seguridad de su aplicación en humanos y la pauta de administración adecuada.
- Fase II: Si se obtiene razonable evidencia de las Fases I y II, comienza esta fase, donde la participación empieza a incrementarse involucrando múltiples médicos tratando cientos o incluso miles de pacientes. El objetivo es verificar la eficacia del fármaco, observar manifestaciones de toxicidad no detectadas con anterioridad y cuantificar la relación entre seguridad y eficacia contra la afección a tratar. Dura entre dos y tres años y en ella participan alrededor de 200 pacientes, a los que se les divide en dos grupos: unos reciben el fármaco y otro (grupo de control) recibe o un placebo que en caso de fármacos para el cáncer son el mejor medicamento del mercado contra la patología. El objetivo es ratificar la eficacia del fármaco.
- Fase III: Si se obtiene razonable evidencia de las Fases I y II, comienza esta fase, donde la participación empieza a incrementarse involucrando múltiples médicos tratando cientos o incluso miles de pacientes. El objetivo es verificar la eficacia del fármaco, observar manifestaciones de toxicidad no detectadas con anterioridad y cuantificar la relación entre seguridad y eficacia contra la afección a tratar.
- Fase IV: También conocida como estudios de farmacovigilancia, que consiste en el seguimiento del fármaco después de que ha sido comercializado. Se busca básicamente la detección de toxicidad previamente insospechada, así como de la evaluación de la eficacia a largo plazo. También se puede estudiar si el medicamento puede administrarse en dosis diferentes a las aprobadas o de distinta forma (por ejemplo, pasar de ser oral a ser inyectable), incluso, se investiga si puede servir para otra enfermedad. En este caso, se seguirá el proceso de las fases I, II Y III mencionadas.
Regulación de los ensayos clínicos
Regulación de los ensayos clínicos

Los ensayos que se realizan para desarrollar un fármaco están regulados por ley y bajo una serie de principios éticos a respetar y seguir, especialmente en la fase en la que los ensayos implican la participación de personas.
Aspectos legales
Aspectos legales
El máximo organismo en España que se encarga de la autorización de los ensayos clínicos, revisando que se ajusten a la ley, es la Agencia Española de Medicamentos y Productos Sanitarios (AEMPS), perteneciente al Ministerio de Sanidad. Este organismo es el responsable de las actividades de evaluación, autorización, registro y control de medicamentos de uso humano en nuestro país.
Además, la Ley del Medicamento española protege a los participantes de los ensayos clínicos al establecer que deben realizarse en condiciones de respeto a sus derechos fundamentales como persona y siguiendo unos principios éticos fijados a nivel mundial en la Declaración de Helsinki (1964) y posteriores revisiones.
A nivel europeo, existe el Reglamento (UE) n °536/2014 del Parlamento Europeo y del Consejo sobre las disposiciones legales, reglamentarias y administrativas de los Estados miembros sobre la aplicación de buenas prácticas clínicas en la realización de ensayos clínicos de medicamentos de uso humano. Esta Directiva se ha ido incorporando desde entonces a la legislación de cada país.
Aspectos éticos
Aspectos éticos
En primer lugar, hay que dejar claro que el ordenamiento jurídico internacional nos protege para que la normativa a aplicar en materia de ensayos clínicos sea estricta, se cumpla a todos los niveles y que, las agencias reguladoras y otros organismos independientes igualmente regulados, velen porque así sea.
Estos principios éticos han sido paulatinamente asumidos por las autoridades sanitarias de los países, incluyéndolos junto a las normativas legales. Algunos de esos principios son que la población sobre la que se va a realizar los ensayos clínicos sean también la beneficiaria de los resultados, la obligación de publicar los resultados si son negativos o la transparencia e información sobre las fuentes de financiación.
Por eso, antes de comenzar la investigación clínica de un nuevo medicamento hay que valorar si el beneficio colectivo potencial de la investigación compensa el riesgo que supone dicha investigación para las personas implicadas en ella. Esa necesidad de proteger a las personas ha llevado al desarrollo de una serie de recomendaciones a seguir durante el proceso de investigación y desarrollo de un fármaco:
- Respeto por las personas, que se traduce en que participen en la investigación voluntariamente y estén adecuadamente informadas. La aplicación práctica de este principio ético es el “Consentimiento Informado”, un procedimiento por el que recibimos información veraz del investigador del ensayo, se nos permite el tiempo suficiente para la compresión del mismo antes de tomar cualquier decisión, la cual debemos expresar libre y voluntariamente. La información que recibimos al participar deberá incluir tanto los beneficios como los riesgos, los objetivos del ensayo, el tratamiento que puede sernos administrado, además de los derechos que nos asisten de privacidad de datos, posibilidad de retirada en cualquier momento, compensación económica y tratamiento en caso de daño o lesión. Dicha información estará recogida en un documento por escrito.
- Asegurar el bienestar de las personas que participan, es decir, no causando daño, extremando los posibles beneficios de la investigación y minimizando los posibles riesgos.
- Todos los participantes en la investigación tenemos las mismas oportunidades de recibir el tratamiento que se está ensayando. Esto significa que los participantes formaremos parte de un grupo u otro del ensayo, por ejemplo, en los ensayos randomizados y con grupo de control, mediante asignación aleatoria, teniendo igual probabilidades estadísticas de pertenecer a un grupo como al otro.
Todas estas recomendaciones éticas, son tenidas en cuenta por el Comité Ético de Investigación Clínica. La existencia de este Comité, que es un organismo independiente, es obligatoria para la realización del estudio y son los que vigilan que se respeten los derechos de los pacientes, así como que se ajuste el ensayo clínico a los requisitos legales y éticos fijados por ley.
Protección de datos
Protección de datos
Durante un ensayo clínico se guarda la más estricta confidencialidad de los datos de los participantes para que no se viole su derecho a la intimidad personal. Por ello, los datos recogidos de cada participante se guardarán sin que se le identifique por el nombre y su identidad no será revelada. Además, estos datos serán utilizados solo con los fines del estudio y el personal participante en los ensayos clínicos tendrán que proteger la confidencialidad de dichos datos.
Los participantes podremos acceder a la información recogida a través de nuestro médico y pedir que se corrija cualquier error en la misma. Si los resultados del estudio se publican, su identidad será también confidencial.
El tratamiento de los datos está protegido por la Ley Orgánica 5/1992, de Regulación del tratamiento automatizado de los datos de carácter personal, en especial en lo que al consentimiento del afectado se refiere y como medida para evitar el acceso de personas no autorizadas. Asimismo, a nivel europeo, está el Reglamento General de Protección de Datos (UE) 2016/679.
Responsabilidad civil
Responsabilidad civil
La contratación de un seguro de responsabilidad civil es obligatoria por parte del promotor para cubrir los posibles daños que puedan sufrir las personas a raíz de su participación en un ensayo clínico. Este seguro cubrirá al promotor, al investigador y sus colaboradores, así como al titular del centro o centros donde se realice el ensayo ante las posibles responsabilidades que surjan si hay un daño en la salud del paciente durante el ensayo y hasta un año después de la terminación de este.
Registro e información sobre los ensayos clínicos
Registro e información sobre los ensayos clínicos
La Declaración de Helsinki insta a que “se debe registrar cada ensayo clínico en una base de datos de acceso público antes de reclutar el primer sujeto”.
Además, se considera internacionalmente que la publicación del resultado de las investigaciones en humanos –tanto positivo como negativo- es una obligación científica, ética y moral.
Por ello, cada vez más existen registros públicos de los ensayos clínicos que se llevan a cabo desde entidades públicas, así como privadas. Uno de los registros más conocido es el ClinicalTrials (www.clinicaltrials.gov), un servicio del Instituto Nacional de Salud de los Estados Unidos, que cuenta también con una base de datos con los resultados de estudios clínicos con humanos desarrollados con el apoyo de organizaciones públicas y privadas alrededor del mundo.
Estos registros y bases de datos sobre ensayos clínicos ayudan a los pacientes y los profesionales sanitarios a conocer y a tener acceso a los ensayos clínicos que se están llevando a cabo con tratamientos experimentales. Además, también sirven para analizar los resultados de ensayos clínicos previos, así como tener conocimiento y poder seguir los resultados de los que se están realizando.
Registro Español de estudios clínicos (REec)
Registro Español de estudios clínicos (REec)
Las autoridades sanitarias españolas pusieron en funcionamiento en 2013 el Registro Español de estudios clínicos (REec) que permite saber, tanto a pacientes como a profesionales de la salud, las investigaciones que se están realizando en nuestro país en relación con su enfermedad, o con uno o varios medicamentos determinados.
Por ello cualquiera tenemos acceso libre a este Registro, sin restricción ninguna, a través de la plataforma desarrollada por la Agencia Española de Medicamentos y Productos Sanitarios https://reec.aemps.es/reec/public/web.html
Con esta medida se quiere cumplir con las garantías de transparencia de los medicamentos, fijadas en la Ley 29/2006, así como seguir las recomendaciones de la Declaración de Helsinki.
Este Registro incluye todos los ensayos clínicos autorizados en nuestro país desde su puesta en funcionamiento, además de estudios observacionales autorizados y otros estudios registrables con carácter voluntario que cuenten con el dictamen favorable de un Comité de Ética de la Investigación.
La finalidad de su desarrollo, según la Agencia Española de Medicamentos y Productos Sanitarios, entidad de la que depende el REec, es:
- Garantizar que las decisiones relacionadas con la salud y cuidados médicos se toman con la garantía o aval de datos científicos públicos y, por tanto, reconocidos.
- Garantizar que se ponen a disposición de la sociedad datos y resultados tanto positivos como negativos de investigaciones.
- Proporcionar a los sujetos participantes en dichas investigaciones información previa de calidad.
- Evitar estudios repetitivos o no aceptables, especialmente en niños, ancianos y otras poblaciones vulnerables, potencialmente desfavorecidas o con dificultades para poder tomar una decisión por sí mismos.
- Detectar aspectos científicos poco investigados y facilitar cubrir esas carencias.
- Facilitar la participación en investigaciones recién autorizadas o en marcha para poder así alcanzar resultados fiables.
Beneficios de formar parte de un ensayo clínico
Beneficios de formar parte de un ensayo clínico

Existen muchas razones por las que las personas accedemos a participar en un ensayo clínico. Lo importante es que entendamos el ensayo y demos nuestro consentimiento para participar, cualesquiera que sean nuestras razones.
Cualquiera que sea la razón por la que estemos considerando participar en un ensayo clínico, es importante que antes hablemos con nuestro médico o equipo de atención médica, para comentar los posibles riesgos y beneficios, y para cerciorarnos de que entendemos lo que implica el ensayo. Esta conversación puede tener lugar en varias citas, y podría incluir hablar sobre el formulario de consentimiento informado, que es el documento oficial que firmaríamos si decidimos participar en un ensayo clínico. El formulario de consentimiento informado puede ser bastante largo, ya que es importante que toda la información sobre el ensayo se explique con claridad. Usted tendrá la oportunidad de leer el documento detenidamente, comentarlo con su médico, y tomarse algo de tiempo para sopesar su decisión antes de firmarlo.
Ayudar a futuros pacientes – Las personas accedemos a participar en un ensayo clínico para contribuir al avance de la medicina. Con el tiempo, el impacto que los ensayos clínicos tienen en las vidas de los pacientes es considerable, y a algunas personas nos gusta saber que de su enfermedad puede salir algo positivo.
Beneficio personal – También decidimos participar en un ensayo clínico con la esperanza de lograr un beneficio personal. Algunos pacientes albergan la esperanza de que el fármaco nuevo pueda ayudarles si se han intentado todas las demás opciones. Otras personas participan para aprender más sobre su enfermedad o porque creen que el ensayo les permitirá acceder más rápidamente a atención médica especializada.
Es importante que sopesemos los posibles riesgos de tomar un fármaco nuevo, así como los posibles beneficios. No existe garantía alguna de que el fármaco nuevo o la nueva combinación de tratamientos será mejor que el tratamiento de referencia, y los efectos secundarios podrían ser mucho peores. Un ensayo clínico se lleva a cabo para encontrar respuestas, como si un fármaco nuevo es mejor o más seguro, de manera que hay preguntas que no se podrán contestar hasta que el ensayo haya concluido.
Asimismo:
- Puede suponer la estabilización y/o la curación de la enfermedad.
- Puede suponer vivir y disfrutar de una vida que otros tratamientos convencionales no garantizan.
- Ayuda a implementar ensayos menos agresivos para el bienestar de las personas pacientes.
- Participar en la mejora, la evolución y la innovación en los fármacos.
Ensayos clínicos actuales
Ensayos clínicos actuales
Todos los ensayos clínicos que actualmente se están realizando en España se pueden consultar en el Registro Español de Ensayos Clínicos (REec), página web del Ministerio de Sanidad, Consumo y Bienestar Social.
Aun así, recuerda que la participación queda restringida al cumplimiento de ciertos requisitos, y que en el supuesto de que se accediera, no hay garantías de que nos podamos beneficiar personalmente de la intervención recibida. Por ello. El mejor consejo que te podemos dar siempre es que consultes con el equipo que lleva tu caso.
Información de interés
Información de interés
Soy un paciente: qué debo saber para participar
Soy un paciente: qué debo saber para participar
Los pacientes somos la esencia de los ensayos clínicos. De hecho, no habría ninguna razón de plantear una investigación si no tuviéramos una necesidad pendiente que resolver: supervivencia, calidad de vida, sintomatología o, incluso, la reducción de los efectos secundarios de los medicamentos actuales; porque no todo es la eficacia.
Los hallazgos que se persiguen descubrir son para nosotros, para que vivamos más y mejor. Por eso somos imprescindibles durante el proceso de investigación, desarrollo y aprobación de los fármacos. En un primer momento porque es nuestra obligación validar la relevancia de los ensayos y lo que significarían que tuvieran éxito desde nuestro punto de vista. Pero es que, además, hay que recordar que no se nos puede recetar algo a los destinatarios finales sin haberse confirmado previamente que esta acción será positiva. Es decir, sin que tengamos en la medida de lo posible, la certeza de que los beneficios supera los riesgos que tendríamos que asumir. Un conocimiento, sin embargo, que nunca se conseguiría salvo que haya pacientes dispuestos a recibirlos cuando todavía se evalúa la seguridad y eficacia en los ensayos clínicos. Todo un problema circular, que si bien, se resuelve porque el acceso temprano a nuevas moléculas ofrece la posibilidad de ampliar el número de alternativas terapéuticas, innovadoras, para tratar el cáncer de pulmón, mucho antes de que salgan al mercado. Y, por supuesto, la satisfacción personal de colaborar con un descubrimiento que mejora la salud de muchos más pacientes.
No obstante, e independientemente de las razones que pueden llevar a un paciente a interesarse por participar en un ensayo clínico y, antes de decidirse, es ineludible informarse sobre:
- Los beneficios potenciales.
- Los posibles riesgos.
- Conocer nuestros derechos.
- Saber todos los detalles de lo que sucederá durante nuestra participación en el ensayo clínico.
Desde aquí despejamos algunas dudas frecuentes que te pueden surgir al plantearte si participar.
¿Quién puede participar?
¿Quién puede participar?
Los ensayos clínicos incluyen a personas afectadas de las enfermedades que son objeto de estudio y, de manera muy específica, a voluntarios sanas en la Fase I. Aunque cuando se trata de fármacos para el cáncer, no se investiga con personas sanas.
Para poder participar, los pacientes debemos que cumplir los criterios de inclusión fijados durante la fase de diseño del ensaño. Es necesario cumplir estos criterios que son habitualmente de edad, sexo, etapa de la enfermedad, tratamientos seguidos anteriormente, etc. Los investigadores utilizan estos criterios para identificar a los participantes más adecuados.
¿Qué debo hacer antes de considerar mi participación?
¿Qué debo hacer antes de considerar mi participación?
La decisión de inscribirse en un ensayo clínico es muy personal y solo podemos tomarla el propio interesado. Si estuvieras en esta situación, antes de decidir, asegúrate de conocer y entender cuáles son los pros y los contras de participar: información que puedes consultar en la hoja informativa en la que se explican en detalle los posibles beneficios y riesgos de participar en el ensayo. Si lo haces, es posible además que te surjan dudas, pero lo más común es que te ofrezcan también una reunión conjunta para resolverlas con el médico que te propuso el ensayo y con el investigador principal del mismo.
Una vez recibas la información y hayas reflexionado sobre ella, si optas finalmente por aceptar, tendrás que firmar el “consentimiento informado”: el documento en el que se vuelve a detallar las características del ensayo para que puedas expresar que has tomado la decisión libre y voluntariamente después de haber entendido la información facilitada. Es así como se garantiza que nadie se inscribe sin haber recibido la información necesaria.
¿Es voluntaria la participación?
¿Es voluntaria la participación?
Sí, la participación en un ensayo clínico siempre es voluntaria. Y el “consentimiento informado” es el documento que lo constata.
¿Me puedo retirar?
¿Me puedo retirar?
Sí, todos los participantes de un ensayo clínico pueden retirarse en cualquier momento. Para ello, manifestarán su decisión de revocar el consentimiento informado que en su momento otorgaron. Además, de esta decisión no se derivará ninguna responsabilidad ni perjuicio alguno.
¿Se recibe alguna compensación económica?
¿Se recibe alguna compensación económica?
En general, no. Aunque se pueden cubrir los costes (traslado, alojamiento, etc) derivados de la participación en el ensayo clínico.
¿Cuáles son los riesgos? ¿Y los beneficios?
¿Cuáles son los riesgos? ¿Y los beneficios?
Los riesgos y beneficios específicos de participar en un ensayo concreto se explican antes de que se acepte participar.
A grandes rasgos, un primer beneficio es tener acceso a nuevos tratamientos médicos para la enfermedad que se investiga, recibiendo además más atención del equipo de investigación que la que podría recibirse fuera del mismo. Además, se tiene un papel más activo en el cuidado de la propia salud. Por último, un aspecto también importante es que con tu colaboración ayudarás a contribuir al progreso médico en beneficio de la sociedad en general.
En cuanto a los riesgos de los ensayos clínicos, hay que destacar que el tratamiento puede que no sea eficaz y que aparezcan efectos secundarios desagradables e incluso graves. Por otro lado, también hay que ser conscientes de que deberá dedicar tiempo a las visitas, estancias en el hospital, exámenes médicos y pruebas.
¿Son seguros? ¿Cuál es mi protección?
¿Son seguros? ¿Cuál es mi protección?
La participación de personas en los ensayos clínicos está protegida con estrictas normas éticas y la legislación vigente.
En la práctica esta protección se basa en cuatro procedimientos de obligado cumplimiento:
- Aprobación del protocolo por el Comité Ético de Investigación Clínica.
- Otorgamiento del consentimiento informado.
- Custodia segura de los datos médicos y personales
- Cobertura de daños: el promotor del ensayo clínico está obligado a la firma de un seguro para cubrir a los pacientes en caso de efectos adversos y daño en su salud.